Poster Presentation The Joint Annual Scientific Meetings of the Endocrine Society of Australia and the Society for Reproductive Biology 2017
An unusual cause of headaches in an adolescent (#275)
Introduction
This is a case of hypophysitis in an adolescent woman whose histological diagnosis is contentious and whose long-term monitoring plan is unchartered territory.
Case Presentation
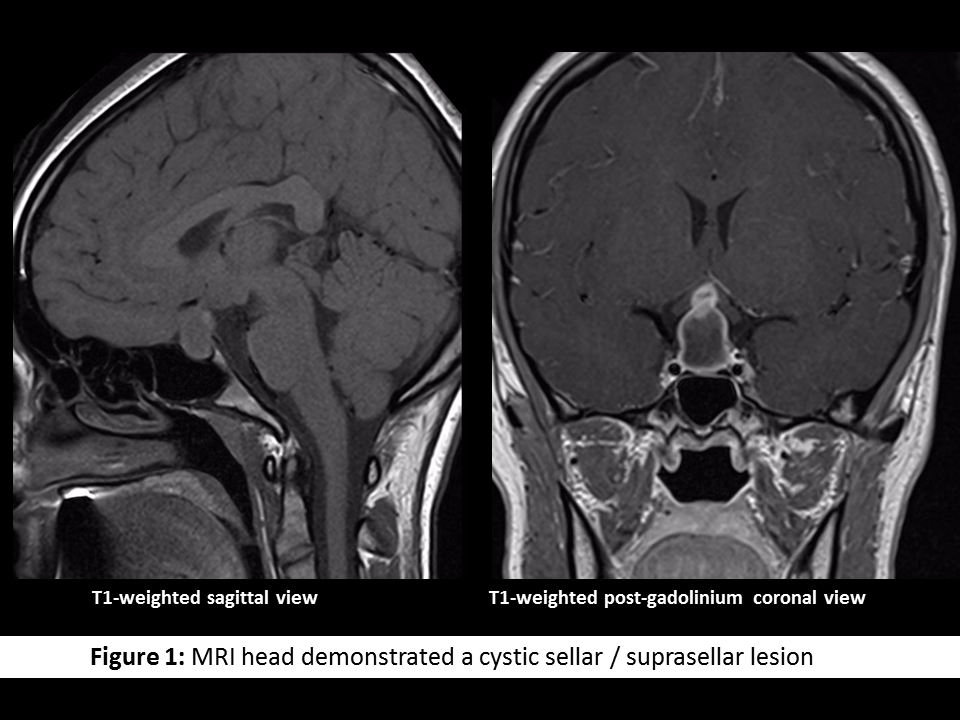
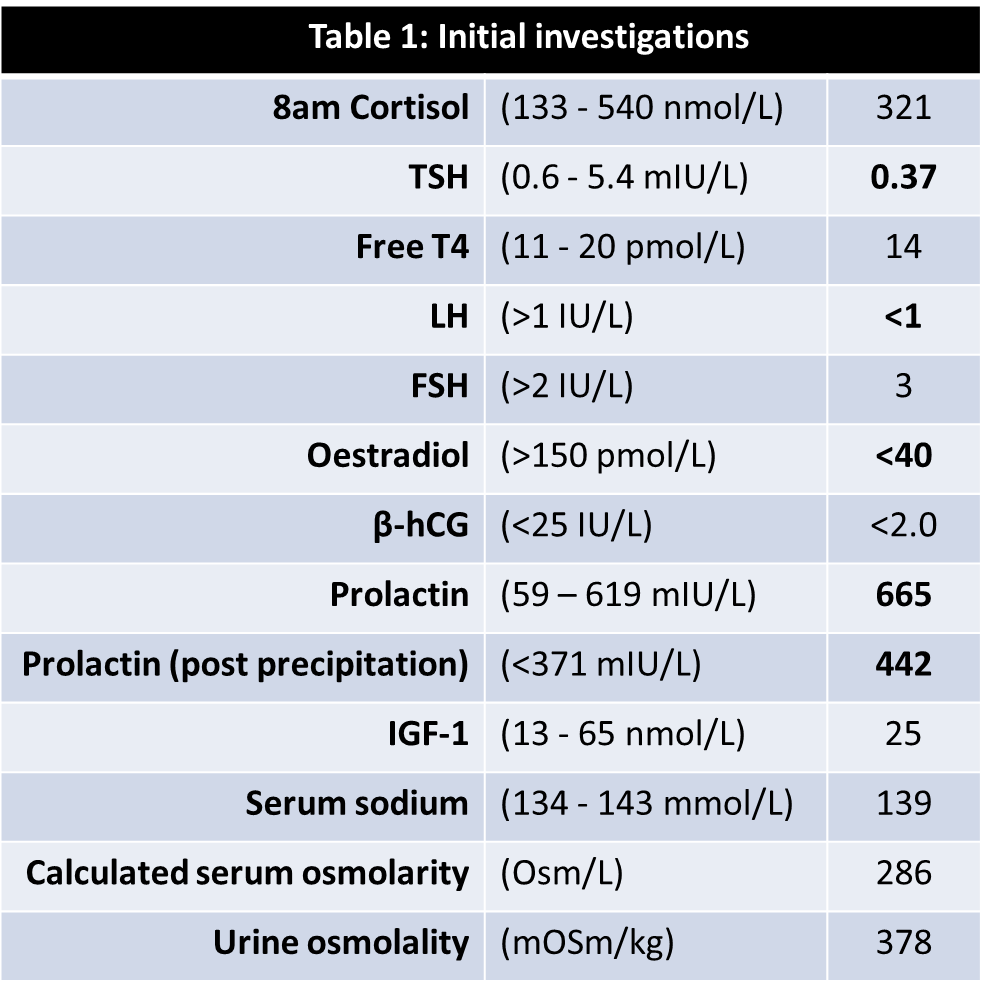
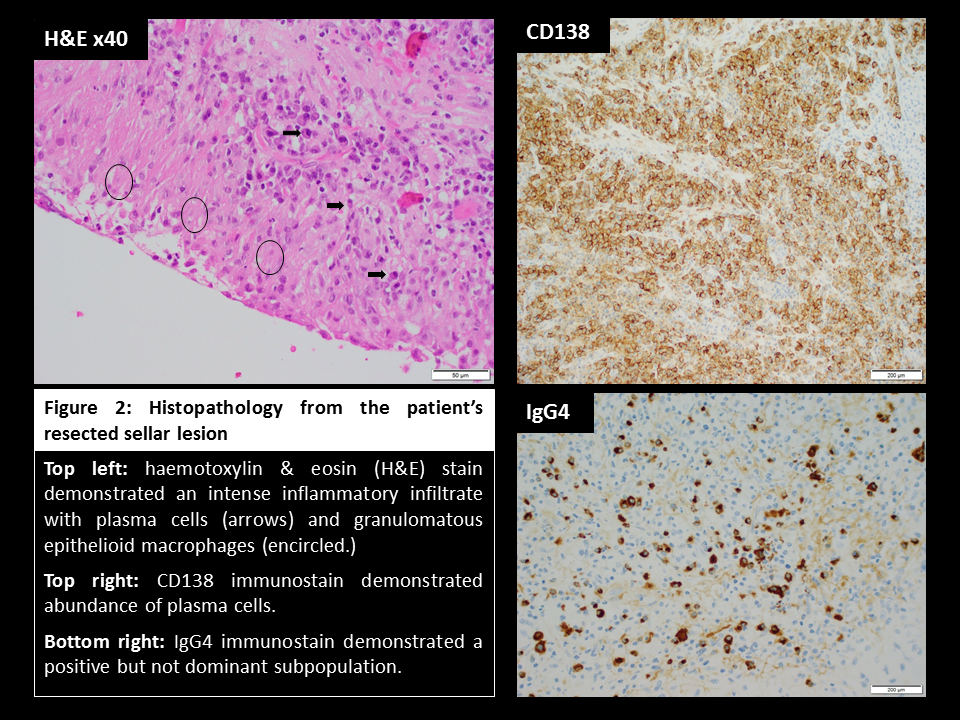
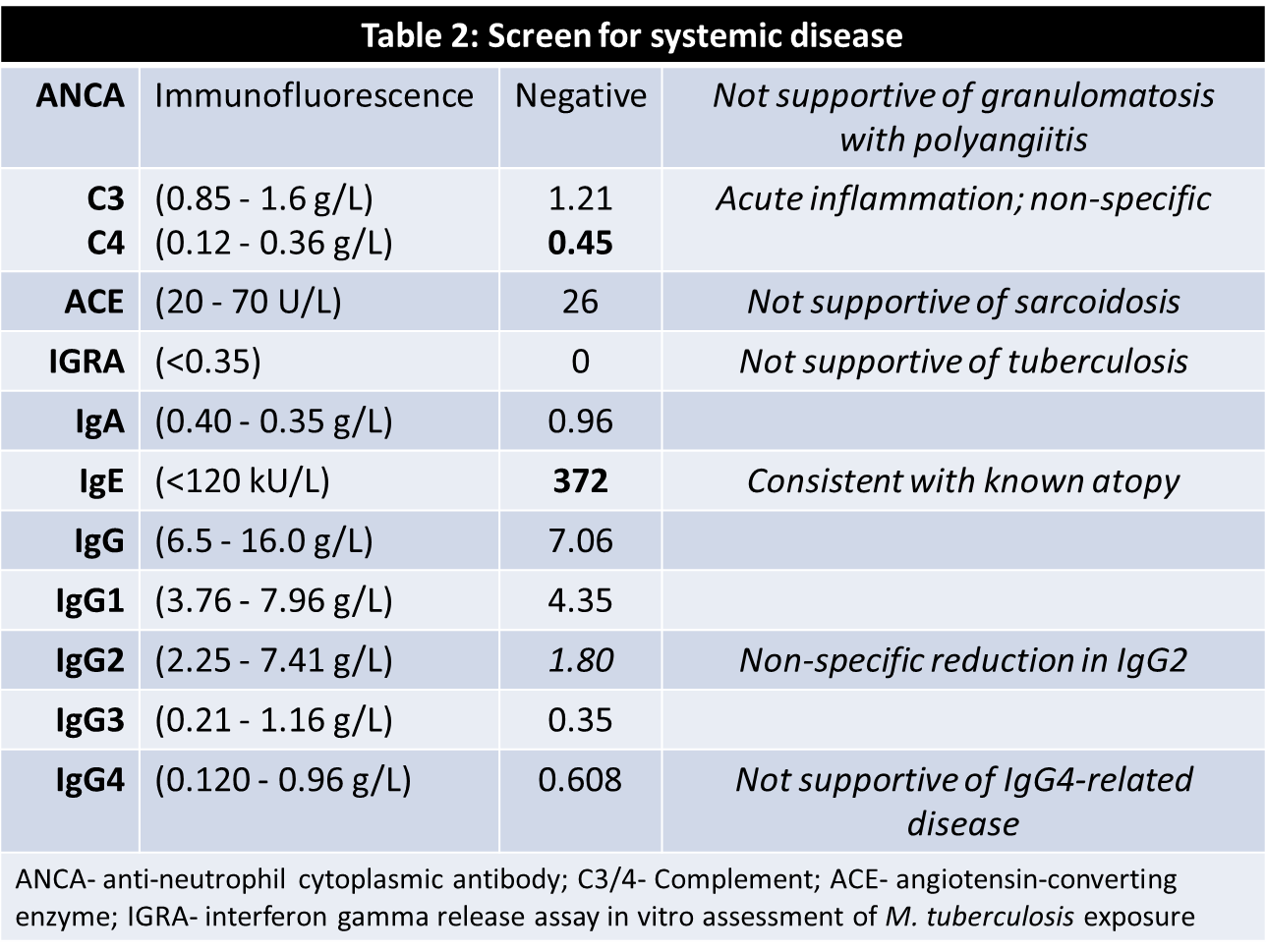
A 16-year-old female presented with a listless conscious state and a 10-week history of headaches, nausea, secondary amenorrhoea, polydipsia and polyuria. Examination revealed bitemporal hemianopia. Pituitary MRI (Figure 1) identified a cystic lesion in the sella turcica with a cystic suprasellar component encroaching upon the optic chiasm. Blood tests demonstrated hypogonadotropic hypogonadism, suppressed thyrotropin, and a mildly elevated prolactin (Table 1). The preliminary diagnosis was cystic craniopharyngioma and the patient proceeded to trans-sphenoidal resection. Histological assessment of sellar tissue did not identify any features of craniopharyngioma. Instead, a florid inflammatory process with IgG4-immunopositive cells and granulomatous reaction was apparent (Figure 2). Investigation for systemic disease with blood tests (Table 2), CT and PET imaging did not further the diagnosis.
Discussion
Hypophysitis is an inflammation of the pituitary gland that gives rise to a non-secretory sellar mass.1 Traditionally, distinct histological variants have been described but recently it has been proposed that some histological features represent the spectrum of hypophysitis progressing over time.2 Both IgG4 plasmacytic and granulomatous hypophysitides are rare, and not described in the paediatric population, bar one other case report.3 Both may occur in isolation but can also precede or follow the diagnosis of other organ involvement. Immune-directed therapy, e.g. glucocorticoids or rituximab, may obviate the need for surgical intervention when hypophysitis is one feature of widespread disease.
Conclusions
The histological features of IgG4 plasmacytic hypophysitis and granulomatous hypophysitis are likely incompletely described and defined, owing to the current paucity and heterogeneity of cases. With equivocation over the underlying cause in our patient, and in the absence of known persistent or systemic disease, immunosuppressive therapy was not provided; however, ongoing surveillance remains necessary.
- Leporati P, Landek-Salgado MA, Lupi I, et al. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum J Clin Endocrinol Metab 2011;96(7):1971-80
- Hunn BHM, Martin WG, Simpson Jr S et al. Idiopathic granulomatous hypophysitis: a systematic review of 82 cases in the literature Pituitary 2014;17:357-65
- Decker L, Crawford AM, Lorenzo G et al. IgG4-related hypophysitis: case report and literature review Cureus 2016;8(12):e907